Fcc Si 优化晶格常数
这是按照VASP官方wiki的这篇教程进行的:
https://cms.mpi.univie.ac.at/wiki/index.php/Fcc_Si
##任务
针对 fcc 结构的硅晶体,用VASP做晶格常数优化
Lattice constant optimization for fcc Si.
输入文件
POSCAR
表示晶格的几何结构参数和位置信息。
1 | fcc Si: |
第一行是注释,可以写体系名称。
第二行是晶格尺寸放大因子,并不是一般意义的晶格常数。
第三行到第五行是晶格的三个基矢的坐标表示。
第六行是一个原胞中每种原子的个数,fcc Si 体系只有一种原子,故写 1。
第七行表示选择 Cartesian 模式,也就是接下来的原子坐标是相对值,实际值应该是相对值乘以晶格常数。如果是 direct coordinates (respectively fractional coordinates) 模式,也就是相对分数坐标,这里是以晶格基矢作为单位长度。如下图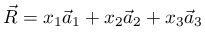
第八行表示一个晶胞中,硅原子的位置相对坐标。
INCAR
VASP最重要的输入文件,决定如何进行计算,包含诸多计算设置参数
1 | System = fcc Si |
KPOINTS
1 | k-points |
POTCAR
POT这部分我不懂,我用的是这个文件夹里的Si的POTCAR,先解压,然后在挪到 node0 上面
1 | zcat /share/apps/src/vasp/pseudopotential/pot/Si/POTCAR.Z > POTCAR |
跑计算
将以下代码复制保存到 loop.sh 脚本,并将 BIN = xxx 那一行改成服务器上VASP的位置,用 qsub loop.sh 在服务器上提交任务。
1 | #! /bin/bash |
执行之后输出
1 | 3.5 1 F= -.51818779E+01 E0= -.51796045E+01 d E =-.454679E-02 |
也就是我们依次将晶格常数设置成3.5到4.3中的值,然后跑VASP计算,并且将计算结果中的能量值挑出来画图。我在OUTCAR里找到对应的能量值,发现在文档最后面。
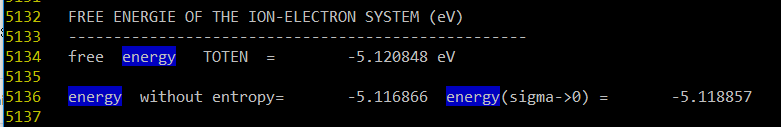
另外我还找到了 energy-cutoff 等于240.00 eV,不知道为啥我在 POTCAR 里没找到
作图
用 gnuplot 作图,可能先要下载 gnuplot,然后开启服务器图形功能, 通过ssh -X -Y 登录主机。
1 | gnuplot |
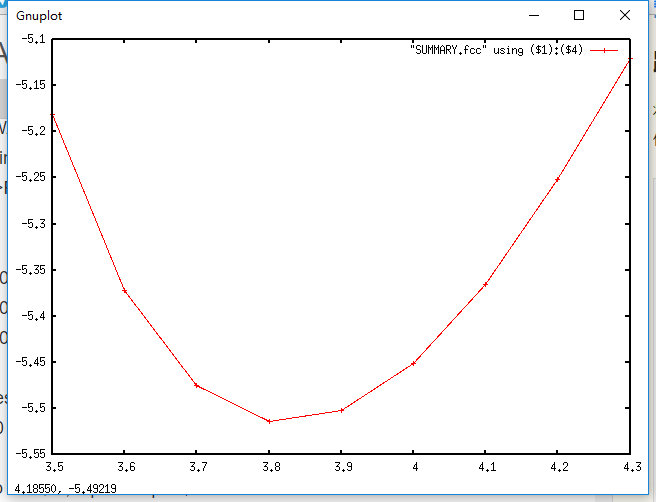
可见晶格常数为3.8 A 时,体系能量最低。
