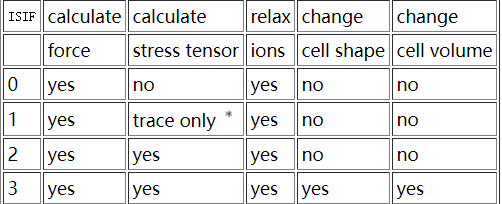
a = [5.6, 5.8, 6.0, 6.2, 6.4, 6.8, 6.9, 7.0, 7.1, 7.2, 7.4] with open("SUMMARY.txt", "wt") as f: for i in a: with open("%.1f/OSZICAR"%i) as fin: for line in fin: pass print(i,line[:-1], file=f)
得到 SUMMARY.txt 如下,第一列是晶格常数
1 2 3 4 5 6 7 8 9 10 11
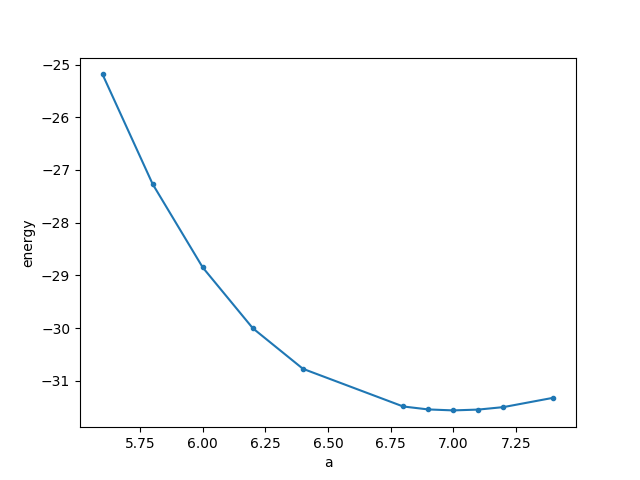
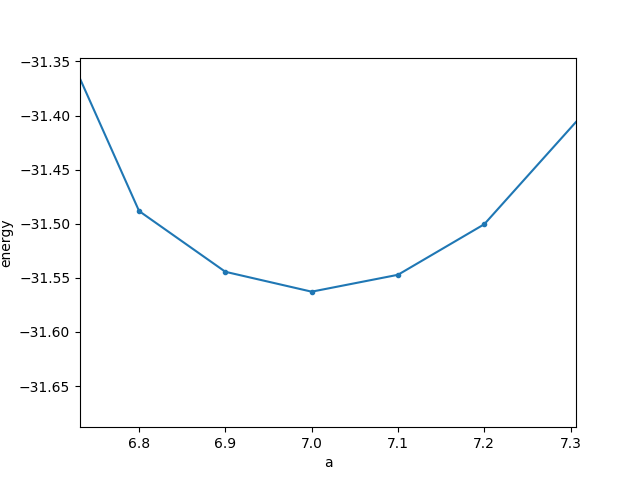
5.6 10 F= -.25231765E+02 E0= -.25185740E+02 d E =-.829857E-03 mag= 5.3969 5.8 12 F= -.27302282E+02 E0= -.27267145E+02 d E =-.256012E-03 mag= 6.0472 6.0 11 F= -.28870757E+02 E0= -.28850240E+02 d E =-.123916E-04 mag= 6.0440 6.2 10 F= -.30009049E+02 E0= -.30004278E+02 d E =0.422491E-05 mag= 6.0047 6.4 13 F= -.30774062E+02 E0= -.30773558E+02 d E =-.483616E-05 mag= 6.0001 6.8 9 F= -.31488252E+02 E0= -.31488244E+02 d E =-.170946E-03 mag= 6.0000 6.9 9 F= -.31544414E+02 E0= -.31544408E+02 d E =-.308623E-03 mag= 6.0000 7.0 11 F= -.31562794E+02 E0= -.31562788E+02 d E =-.100152E-03 mag= 6.0000 7.1 14 F= -.31547196E+02 E0= -.31547189E+02 d E =-.146476E-04 mag= 6.0000 7.2 10 F= -.31500496E+02 E0= -.31500486E+02 d E =-.181810E-03 mag= 6.0000 7.4 11 F= -.31322697E+02 E0= -.31322675E+02 d E =-.108923E-02 mag= 6.0000
再写一个作图脚本 plot.py
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
import matplotlib.pyplot as plt
a = [] energy = [] with open("SUMMARY.txt", "r") as fin: for line in fin: mylist = line.split() a.append(mylist[0]) energy.append(mylist[5])